Eye diseases
Retinitis pigmentosa
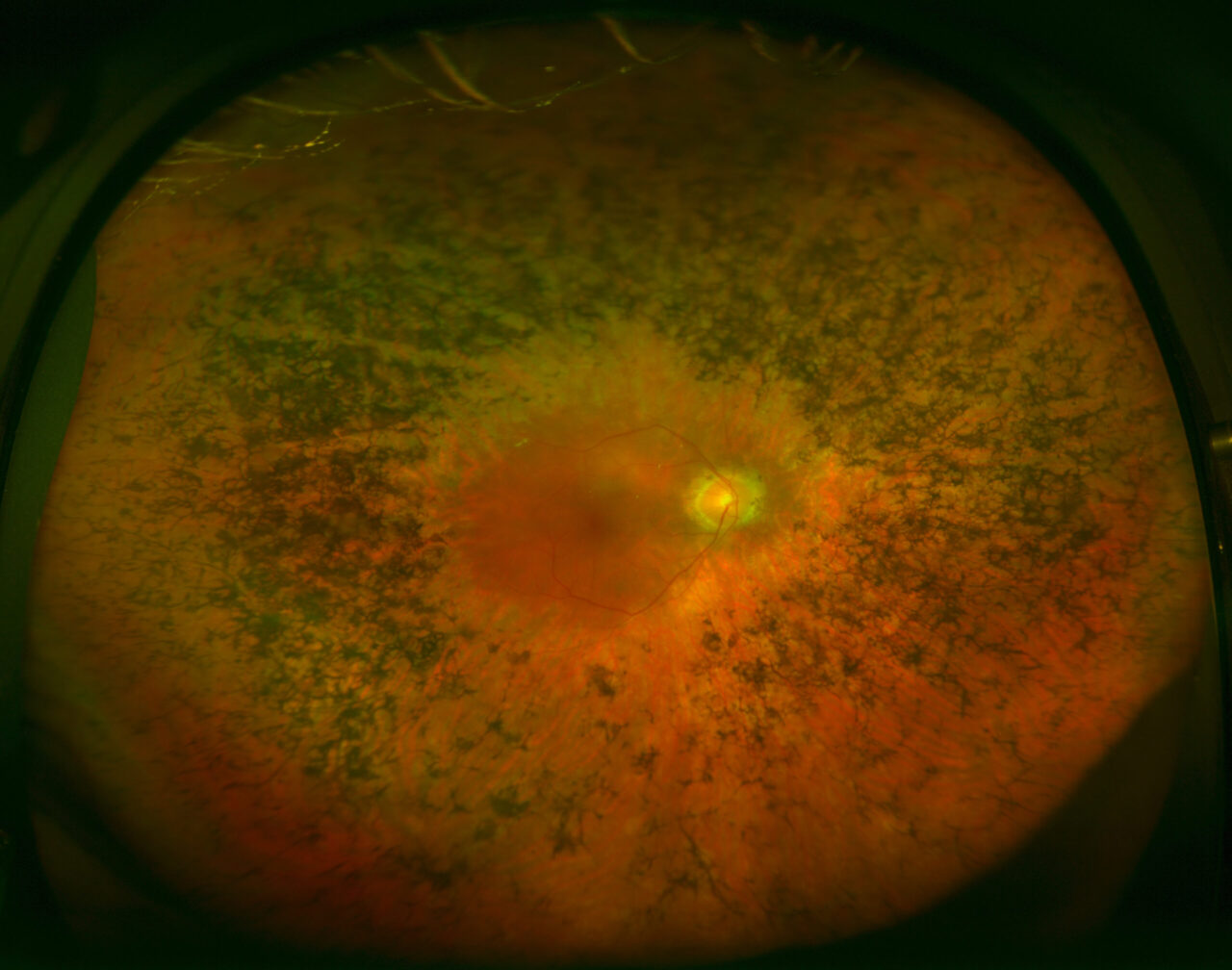
What is retinisis pigmentosa?
Retinisis pigmentosa is a hereditary dystrophy of the retina (link), classed as a “rare” disease that affects 2 or 3 out of 10,000 people and is the main cause of genetic blindness in the adult population.
This disease progressively damages the retina (the innermost layer of the eye), leading to the loss or “programmed cell death” of its photoreceptor cells. These are responsible for turning the light stimuli entering the eye into nerve or electric signals that are sent to the brain. Retinisis pigmentosa typically affects the photoreceptors known as rods (responsible for peripheral vision) and respects the cones (responsible for central vision). However, general atrophy of the retina might occur during final stages.
Symptoms
Causes and risk factors
Diagnosis
Treatment
Despite being born with the disease, retinisis pigmentosa does not often become apparent until adolescence or young adulthood, and the age at which it appears varies. The first signs are normally poor night vision (failure to adapt to the dark) and the progressive reduction of the visual field. As the disease progresses, flashes are often seen and, in more advanced stages, a decrease in visual acuity and altered perception of colours.
