
Decoded the mechanism of a mutation that alters chlorine levels in the retina
IMO Grupo Miranza’s research team has made a leap forward in understanding the mechanism behind one of the most prevalent macular dystrophies, Best’s disease. In the course of an IMO Foundation study on the effect of genetic mutations in seven patients with retinal dystrophies (Stargardt, retinitis pigmentosa, achromatopsia and Best’s disease), researchers have come up with this important finding, which was recently published in the International Journal of Molecular Science.
The patients included in the study, which began five years ago, underwent a skin biopsy to induce stem cells, which have now been converted into retinal cells using cell differentiation techniques to study why each patient’s genetic mutation causes their retinal dystrophy.
A breakthrough in macular dystrophies with added value
In this research, researchers have studied, among other things, a particular mutation, which is not described in other families, in a patient with Best’s disease, the second most prevalent macular dystrophy, which usually appears in childhood or adolescence and affects the macula, while sharply reducing visual acuity. The gene involved in the disease is the BEST1 gene, for which more than 350 mutations have been described, but none, so far, like the one in this patient. Moreover, the fact that in this case the research was carried out using the patient’s own cells, instead of using in vitro models, as in other studies, adds value to the finding.
According to Dr Esther Pomares, Head of IMO Grupo Miranza’s Genetics Department, “we have been able to decipher and describe the specific mechanism that triggers this mutation, altering the entry of chlorine into the cells of the retinal pigment epithelium, which are affected by this disease.”
Specifically, the geneticists involved in the IMO Foundation study have analysed the consequences of the alteration of amino acid 77, where the patient’s mutation occurs. This amino acid is located in the neck of the channel that regulates its opening and closing and the entry of chlorine into the retinal cells. Researchers have been able to figure out that this alteration leaves the channel open, and not closed, as has been described in most previously studied cases of this pathology.
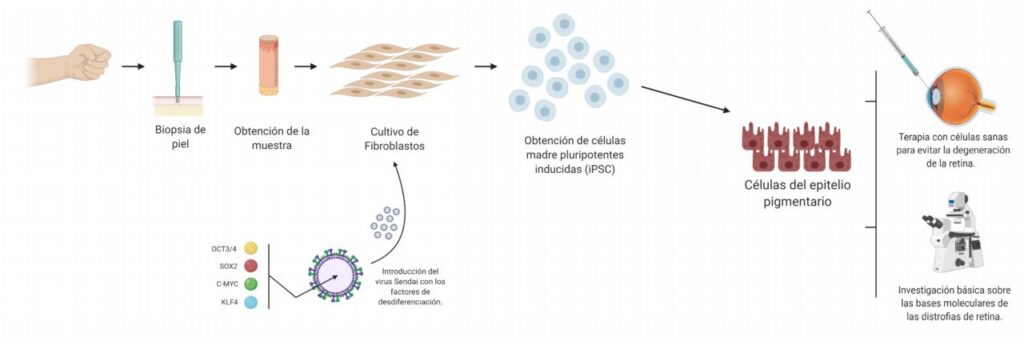
Possibility of specific retinal therapies for each patient
According to Dr Arnau Navinés, the researcher who led the study, “a better understanding of the disease’s mechanism gives way to the design of specific therapies for each patient.” In this case, for instance, “we would consider a blocking or adding gene therapy, cancelling the mutant protein or adding bestrophins (protein of the BEST1 gene), to restore the appropriate levels of this protein. This would prevent cell death and halt the progression of the disease, as well as regulating the levels of chlorine in the retina.”